 作者投稿
作者投稿 专家审稿
专家审稿 编辑办公
编辑办公 邮件订阅
邮件订阅 RSS
RSS
Clinical Characteristics and Genetic Mutations of Chinese Patients with X-linked Retinoschisis
-
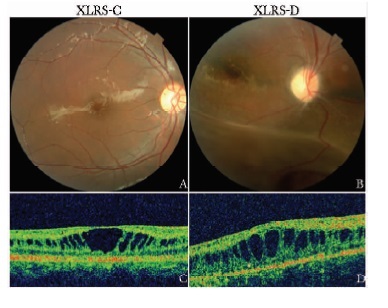
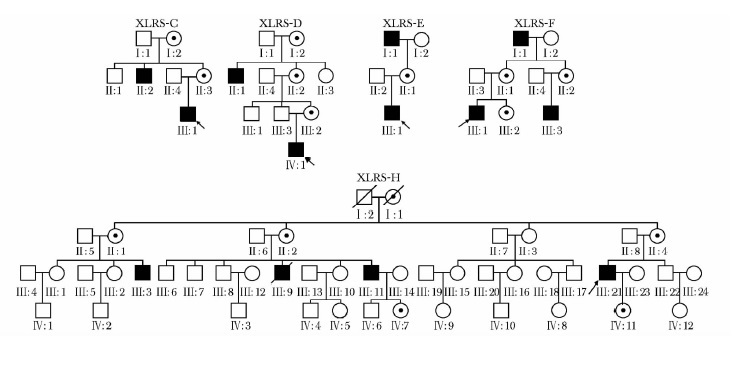
摘要:目的 探讨中国遗传性视网膜劈裂症(X-linked retinoschisis, XLRS)患者的临床特征及致病基因突变位点。方法 研究纳入北京协和医院眼科经临床确诊的5个XLRS家系(包含12例患者)、10例散发病例、16名女性携带者和100名无眼疾的正常对照者。5个家系的先证者及10例散发病例(共15例患者)接受了详细的眼科检查。采集22例患者及16名携带者的外周静脉血3 ml用于提取基因组DNA。采用聚合酶链反应法对RS1基因的6个外显子片段进行扩增后直接测序, 以明确突变位点及突变类型。结果 RS1基因检测共发现11种致病性突变, 包括p.E72K、p.W92C、p.R102Q、p.W112X、p.S134P、p.R156G、p.P193S、p.R200H、p.R209C、p.R213W和p.R213Q, 其中p.W112X和p.S134P突变为新发突变。1例散发病例未确定RS1基因突变位点。在确定突变的所有患者中, 5个家系的先证者和9例散发患者主诉均为不同程度的视力下降, 最佳矫正视力在0.04~0.8之间。眼底检查均存在特征性黄斑区劈裂, 其中有6例患者合并周边视网膜劈裂。携带RS1基因突变的患者中有10例接受视网膜电图检查, 结果显示9例患者的混合反应均呈特征性的负波形。携带同一致病性突变的不同患者病情轻重程度有所不同。结论 XLRS患者在临床表型上存在较大差异, 未观察到基因型与表型间的相关性。Abstract:Objective To assess the clinical features and to identify genetic mutations in Chinese patients with X-linked retinoschisis (XLRS).Methods Patients were recruited from ophthalmic clinics in Peking Union Medical College Hospital. A cohort of five unrelated XLRS families (including 12 XLRS patients), 10 sporadic XLRS patients, 16 female carriers and 100 normal subjects were enrolled in this study. Clinical evaluation was performed on all probands from 5 unrelated families and 10 sporadic cases involved in this study (15 patients in total). Genomic DNA was extracted from the peripheral leukocytes of 22 patients and 16 carriers. All exons and the flanking introns of the RS 1 gene were amplified by polymerase chain reaction and screened for mutations by direct DNA sequencing.Results A total of 11 mutations including p. E72K, p. W92C, p. R102Q, p. W112X, p. S134P, p. R156G, p. P193S, p. R200H, p. R209C, p. R213W and p. R213Q were identified, among which p. W112X and p. S134P were two novel mutations. We did not find any mutation in one sporadic case. Clinical evaluation on patients with RS1 mutation (14 patients in total) showed that reduced visual acuity was the common initial symptom in all 14 patients. The best corrected visual acuity ranged from 0.04 to 0.8. Typical foveal schisis was found in all 14 patients, among whom 6 were found to be with peripheral schisis. Electroretinogram was carried out on 10 patients with RS1 gene mutations and 9 of them displayed electronegative in the standard combined response. Patients with same RS1 mutation exhibited remarkable phenotypic differences.Conclusions Chinese patients with XLRS displayed variability in clinical phenotypes. No association was found between the genotypes and phenotypes.
-
Keywords:
- X-linked retinoschisis /
- RS1 gene /
- mutation /
- phenotype
-
特发性黄斑裂孔(idiopathic macular holes, IMH)是引起老年人视力下降的常见病因, 在中国北方地区40岁以上人群中的发病率为1.6‰[1], 其发病高峰年龄段为60~70岁, 女性多见[2], 主要表现为无明显诱因的黄斑区全层视网膜组织的缺失。虽然目前广泛认可玻璃体黄斑牵拉在IMH的发生中起重要作用, 但是研究发现很多其他因素也参与了IMH的发病过程, 包括退行性黄斑变薄、黄斑囊样变、色素上皮病变以及系统性血管病变等; 尤其是最近有研究发现脉络膜局部血流的改变可能与黄斑部的一些病变有关, 如年龄相关性黄斑变性、中心性浆液性脉络膜视网膜病变、特发性黄斑前膜及IMH等[3-5]。
光学相干断层扫描(optical coherence tomography, OCT)技术在黄斑部疾病诊断方面发挥重要作用, 新一代频域OCT的出现使得对黄斑部的观察更加细致深入。Spaide等[6]运用频域OCT开发出深度增强成像(enhanced depth imaging, EDI)技术, 使得活体观察脉络膜形态及结构成为可能。运用EDI技术的研究发现, 脉络膜厚度与年龄呈负相关, 年龄每增加10岁, 中心凹下脉络膜厚度下降15.6μm[7]。IMH好发于老年人, 脉络膜厚度与IMH发病之间是否存在一定关系, 本研究就此问题进行探讨。
对象和方法
对象
收集2011年6月至2012年12月在北京协和医院眼科就诊的20例单眼IMH患者及同时期于门诊就诊并排除眼部疾患且年龄、性别均匹配的正常对照20名。IMH患者的患眼和对侧健眼均作为研究对象, 正常对照随机选择左眼或右眼作为研究对象。将所有眼分为IMH患眼组20眼, 对侧健眼组20眼, 正常对照组20眼。
排除标准包括:近视超过-3.0D、弱视、青光眼、葡萄膜炎病史、眼部外伤或肿瘤史、视网膜变性类疾病、脉络膜新生血管、中心性浆液性脉络膜视网膜病变、糖尿病视网膜病变、屈光手术史、眼底激光光凝史、服用糖皮质激素、玻璃体腔注射史及屈光介质混浊影响OCT检测等情况。
脉络膜厚度测量方法
所有患者行双眼频域OCT (Heidelberg Spectralis, Heidelberg, Germany)检查, 采用EDI技术, 对后极部黄斑中心凹进行0度和90度扫描, 每张OCT图均由100个扫描图叠加成像。脉络膜厚度的测量方法为色素上皮外界和巩膜内界之间的垂直距离。所有测量由同一位医生完成, 每个点测量3次, 取平均值作为测量数据。比较3组中心凹下脉络膜厚度值(subfoveal choroidal thickness, SFCT)和距中心凹1和2mm距离的鼻侧脉络膜厚度值(nasal choroidal thickness, NCT)、颞侧脉络膜厚度值(temporal choroidal thickness, TCT)、上方脉络膜厚度值(superior choroidal thickness, SCT)和下方脉络膜厚度值(inferior choroidal thickness, ICT)。
统计学处理
应用SPSS 17.0统计软件进行分析。采用单因素方差分析比较3组之间脉络膜厚度, 如果差异具有统计学意义, 进一步采用Tukey-Kramer检验进行多重比较。并对各组研究对象的年龄及SFCT进行相关分析。P < 0.05为差异具有统计学意义。
结果
共有20例IMH患者纳入研究, 均为单眼患病, 包括男性6例, 女性14例, 平均年龄(65.95±5.04)岁。正常对照组共纳入20名, 包括男性6名, 女性14名, 平均年龄(63.50±6.12)岁, 组间的性别及年龄匹配, 差异无统计学意义(P>0.05)。
脉络膜厚度测量结果显示, IMH患眼组、对侧健眼组及正常对照组的SFCT均值分别为(161.53±50.50)、(204.95±59.58)、(248.00±63.40)μm, 3组间差异具有统计学意义(P=0.000), IMH患眼组及健眼组的SFCT均值较正常对照组均显著降低(P < 0.05)。图 1显示了频域OCT测量IMH患眼、对侧健眼及正常对照眼的脉络膜厚度。采用单因素方差分析比较其他8个位点发现, 3组间的脉络膜厚度均值均具有显著差异(P < 0.05)。进一步行Tukey-Kramer检验进行多重比较发现, IMH患眼组各个位点的脉络膜厚度均值较正常对照组显著降低(P < 0.01), 对侧健眼组在除SCT2mm及ICT2mm以外的6个位点较正常对照组显著降低(P < 0.05), 患眼组较对侧健眼组脉络膜厚度值降低, 但差异无统计学意义(P > 0.05)(表 1)。
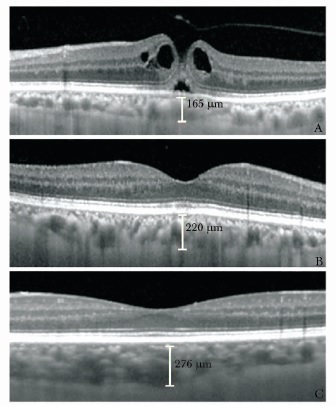
![]() 图 1 频域光学相干断层扫描测量左眼IMH患者(女, 66岁)及正常人(女, 61岁)脉络膜厚度A.IMH患者左侧患眼, SFCT为165μm; B.IMH患者右侧健眼, SFCT为220μm; C.正常对照眼, SFCT为276μm;IMH:特发性黄斑裂孔; SFCT:中心凹下脉络膜厚度值表 1 3组各测量位点脉络膜厚度比较($ \bar x \pm s $,μm)
图 1 频域光学相干断层扫描测量左眼IMH患者(女, 66岁)及正常人(女, 61岁)脉络膜厚度A.IMH患者左侧患眼, SFCT为165μm; B.IMH患者右侧健眼, SFCT为220μm; C.正常对照眼, SFCT为276μm;IMH:特发性黄斑裂孔; SFCT:中心凹下脉络膜厚度值表 1 3组各测量位点脉络膜厚度比较($ \bar x \pm s $,μm)
各组患者年龄与SFCT的相关分析结果显示, IMH患眼组及对侧健眼组的SFCT均值与年龄均无相关性(r=-0.233, P=0.078;r=-0.166, P=0.243), 而正常对照组SFCT与年龄呈负相关(r=-0.291, P=0.024)。
讨论
频域OCT的EDI技术能够活体观察脉络膜全层结构, 一项利用此技术的研究发现SFCT与眼球灌注压相关, 能够间接提示黄斑下方的血流灌注状态[8]。研究发现脉络膜厚度与年龄及眼轴长度呈负相关[9-12]。Margolis等[7]测量得到的正常成人SFCT为287μm, 其与年龄的关系为SFCT=(366-1.56×年龄) μm, 根据此公式计算本研究对象(60~80岁)的SFCT均值约为250μm, 而实际观察得到的正常对照组SFCT均值为248.00μm, 与之基本相符。正常对照组的SFCT均值与年龄呈负相关, 而与之年龄、性别相匹配的IMH患眼组及健眼组的SFCT均值与年龄无相关关系, 提示这两组脉络膜厚度的降低并非由年龄和性别因素引起。
目前广泛认为玻璃体黄斑牵拉是引起黄斑裂孔的主要原因。Gass[13-14]提出除了前后方向的牵拉, 后极部玻璃体皮质对黄斑区切线方向的牵引也是主要病因之一。但是在临床中还观察到一些患者已经发生了完全的玻璃体后脱离或是在玻璃体切除术后, 依然形成黄斑裂孔, 提示除了机械牵拉, 还存在其他因素参与黄斑裂孔的致病过程[15-16]。Aras等[17]利用视网膜血流仪测量黄斑区脉络膜血流流量及速度, 发现4期及1a期黄斑裂孔患者的脉络膜血流量及速度较正常人显著降低。Reibaldi等[18]观察了22例IMH患者, 其患眼的SFCT均值为183.2μm, 对侧健眼的SFCT均值为196.6μm, 正常对照眼的SFCT均值为245.0μm, 患眼及对侧健眼的脉络膜厚度均较正常对照显著降低, 而两者之间差异无统计学意义。本文研究结果与Reibaldi等[18]的结果基本一致, 较其不同之处是增加观察了中心凹上下1mm及2mm处的脉络膜厚度, 结果显示患眼中心凹上下的脉络膜厚度显著降低, 对侧健眼中心凹上下的脉络膜厚度亦有不同程度降低。本研究证明IMH患眼后极部上下鼻颞四个方位的脉络膜厚度普遍明显变薄, 提示脉络膜的改变可能先于黄斑裂孔的发生, 并与其他致病因素如玻璃体牵拉等共同作用, 引起黄斑裂孔。
本研究结果显示IMH对侧健眼脉络膜厚度虽然亦普遍变薄, 但其程度较患眼略轻, 在SCT2mm及ICT2mm两个位点较正常对照组差异无统计学意义, 而曾婧等[19]研究发现对侧健眼脉络膜厚度比正常对照略低, 在各个位点差异均无统计学意义。曾有文献报道, IMH患者对侧健眼有色觉损伤或多焦视网膜电图的下降, 数年后也发生了黄斑裂孔[20-21]。不同研究中对侧健眼脉络膜厚度降低的程度有所不同, 推测可能与各个研究入选患者所处的病程阶段不同有关。但是, 文献报道IMH患者对侧眼产生黄斑裂孔的发病率仅有15%[22], 提示还有其他重要的致病因素参与其中。
Ikuno等[23]研究发现, 视网膜不同位点的脉络膜厚度不同, 中心凹下最厚, 离中心凹越远, 脉络膜越薄, 中心凹下的厚度较鼻颞侧差异显著, 而与上下方的差异并不显著, 颞侧较鼻侧更厚, 上方较下方更厚。本研究的正常对照组也体现了这一规律, 这可能是由于黄斑区的视锥细胞最密集, 代谢最为旺盛, 因此为之提供血流灌注的脉络膜厚度最厚。鼻侧与下方脉络膜厚度更薄可能是脉络膜血管分水岭存在所致。
本研究也存在一定的不足之处,一是缺乏自动测量脉络膜厚度的软件,因此需要操作者人为确定脉络膜的边界,存在一定的测量误差;二是样本量较小,但是组间的显著差异支持了数据的准确性;三是由于黄斑裂孔是进展性疾病,研究者无法得知确切的发病时间。
本研究结果发现IMH患者患眼的脉络膜厚度明显变薄, 对侧健眼的脉络膜厚度亦有不同程度变薄, 提示IMH的发生与脉络膜的血流灌注状态相关, 是一种多因素参与的疾病, 但其确切的发病机制及各因素所发挥的作用还有待进一步研究。
致谢: 衷心感谢参与本次试验研究的所有患者及其亲属的理解及配合。 -
[1] Tantri A, Vrabec TR, Cu-Unjieng A, et al. X-linked retinoschisis:a clinical and molecular genetic review[J]. Surv Ophthalmol, 2004, 49:214-230. DOI: 10.1016/j.survophthal.2003.12.007
[2] George ND, Yates JR, Moore AT. X linked retinoschisis[J]. Br J Ophthalmol, 1995, 79:697-702. DOI: 10.1136/bjo.79.7.697
[3] George ND, Yates JR, Moore AT. Clinical features in affected males with X-linked retinoschisis[J]. Arch Ophthalmol, 1996, 114:274-280. DOI: 10.1001/archopht.1996.01100130270007
[4] Roesch MT, Ewing CC, Gibson AE, et al. The natural history of X-linked retinoschisis[J]. Can J Ophthalmol, 1998, 33:149-158.
[5] George ND, Yates JR, Bradshaw K, et al. Infantile presentation of X linked retinoschisis[J]. Br J Ophthalmol, 1995, 79:653-657. DOI: 10.1136/bjo.79.7.653
[6] Grayson C, Reid SN, Ellis JA, et al. Retinoschisin, the X-linked retinoschisis protein, is a secreted photoreceptor protein, and is expressed and released by Weri-Rb1 cells[J]. Hum Mol Genet, 2000, 9:1873-1879. DOI: 10.1093/hmg/9.12.1873
[7] Ando A, Takahashi K, Sho K, et al. Histopathological findings of X-linked retinoschisis with neovascular glaucoma[J]. Graefes Arch Clin Exp Ophthalmol, 2000, 238:1-7. DOI: 10.1007/s004170050001
[8] Sauer CG, Gehrig A, Warneke-Wittstock R, et al. Positional cloning of the gene associated with X-linked juvenile retinoschisis[J]. Nat Genet, 1997, 17:164-170. DOI: 10.1038/ng1097-164
[9] Molday LL, Wu WW, Molday RS. Retinoschisin (RS1), the protein encoded by the X-linked retinoschisis gene, is anchored to the surface of retinal photoreceptor and bipolar cells through its interactions with a Na/K ATPase-SARM1 complex[J]. J Biol Chem, 2007, 282:32792-32801. DOI: 10.1074/jbc.M706321200
[10] Takada Y, Fariss RN, Muller M, et al. Retinoschisin expression and localization in rodent and human pineal and consequences of mouse RS1 gene knockout[J]. Mol Vis, 2006, 12:1108-1116. https://www.ncbi.nlm.nih.gov/pubmed/17093404
[11] Molday RS. Focus on molecules:retinoschisin (RS1)[J]. Exp Eye Res, 2007, 84:227-228. DOI: 10.1016/j.exer.2005.12.013
[12] Takada Y, Vijayasarathy C, Zeng Y, et al. Synaptic pathology in retinoschisis knockout (Rs1-/y) mouse retina and modification by rAAV-Rs1 gene delivery[J]. Invest Ophthalmol Vis Sci, 2008, 49:3677-3686. DOI: 10.1167/iovs.07-1071
[13] Suganthalakshmi B, Shukla D, Rajendran A, et al. Genetic variations in the hotspot region of RS1 gene in Indian patients with juvenile X-linked retinoschisis[J]. Mol Vis, 2007, 13:611-617.
[14] Vijayasarathy C, Sui R, Zeng Y, et al. Molecular mechanisms leading to null-protein product from retinoschisin (RS1) signal-sequence mutants in X-linked retinoschisis (XLRS) disease[J]. Hum Mutat, 2010, 31:1251-1260. DOI: 10.1002/humu.21350
[15] 余洪华, 李涛, 雷蕾, 等.先天性视网膜劈裂的RS1基因突变型及其与临床表型相关性研究[J].中华眼底病杂志, 2011, 27:409-413. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=zhydb201105003 [16] Forsius H, Krause U, Helve J, et al. Visual acuity in 183 cases of X-chromosomal retinoschisis[J]. Can J Ophthalmol, 1973, 8:385-393. http://europepmc.org/abstract/MED/4742888
[17] Kjellstrom S, Vijayasarathy C, Ponjavic V, et al. Long-term 12 year follow-up of X-linked congenital retinoschisis[J]. Ophthalmic Genet, 2010, 31:114-125. DOI: 10.3109/13816810.2010.482555
[18] Li X, Ma X, Tao Y. Clinical features of X linked juvenile retinoschisis in Chinese families associated with novel mutations in the RS1 gene[J]. Mol Vis, 2007, 13:804-812.
[19] Xu J, Gu H, Ma K, et al. R213W mutation in the retinoschisis 1 gene causes X-linked juvenile retinoschisis in a large Chinese family[J]. Mol Vis, 2010, 16:1593-1600.
-
期刊类型引用(9)
1. 朱海燕,郭菊,周朋义,金波,谢坤鹏,杜利平,金学民. 特发性黄斑裂孔脉络膜厚度及血流灌注观察. 中华眼底病杂志. 2022(09): 755-761 .  百度学术
百度学术
2. 朱海燕,郭菊,周朋义,金波,谢坤鹏,杜利平,金学民. 特发性黄斑裂孔脉络膜厚度及血流灌注观察. 中华眼底病杂志. 2022(09): 755-761 . 百度学术
3. 李思园,赵芃芃,秦梅. 脉络膜厚度及血液循环与特发性黄斑裂孔的相关关系研究现状. 中国眼耳鼻喉科杂志. 2021(03): 221-225 . 百度学术
4. 王丽丽,马玲,柏茂仁,范春宁. 脉络膜厚度与黄斑裂孔的相关性. 国际眼科杂志. 2018(01): 122-125 . 百度学术
5. 李宸宇,周国宏,孔丽,王文娟,贺荣华,王永瑞. 黄斑中心凹下脉络膜血液循环与特发性黄斑裂孔发病关系的研究进展. 世界最新医学信息文摘. 2018(45): 66-67+70 . 百度学术
6. 田雨,郭珊,王松田,喻京生. 脉络膜厚度的临床研究进展. 中华眼科医学杂志(电子版). 2018(04): 181-186 . 百度学术
7. 贺李娴,刘二华. 脉络膜厚度与特发性黄斑裂孔发病关系的研究进展. 国际眼科杂志. 2016(07): 1291-1294 . 百度学术
8. 许静,冯洁,叶青. 特发性黄斑裂孔术中填充物的探讨. 国际眼科杂志. 2016(09): 1746-1749 . 百度学术
9. 郁艳萍,刘武. 脉络膜厚度与特发性黄斑裂孔和黄斑前膜关系的研究进展. 眼科新进展. 2016(09): 898-900 . 百度学术
其他类型引用(12)
 下载:
下载:


计量
- 文章访问数: 218
- HTML全文浏览量: 49
- PDF下载量: 14
- 被引次数: 21












